Tutorial¶
DBGen is a general purpose database to support genomic data anlysis studies. The current implementation is based on mongoDB.
Supported services¶
DBGen has been designed in order to provide user-friendly APIs for typical bioinformatic workflows. The following functionalities are currently supported:
extremely simple interface to the database backend
automatic import of source files containing minimal information about biological samples i.e. project name, run accession, FTP links to download the genotype, and sample phenotypes (if available)
automatic download of sample genotypes (using the provided FTP links)
simple interface to save the results (raw files) of any bioinformatic tool
a collection of methods to query the database efficiently
Backend database¶
Setup¶
In order to use this package, you need to download, install, and configure mongoDB on your machine. You can follow the instructions on the official website.
Start and stop the backend¶
DBGen provides two methods to start and stop the backend database service,
dbgen.start_db and dbgen.shutdown_db:
import dbgen
configs = dbgen.load_cfg()
dbgen.start_db(configs)
...
dbgen.shutdown(configs)
Customize DBGen¶
DBGen can be easily configured using command line parameters. There are 5 user parameters:
--password: user password--database: database name (default:dbgen_test)--host: host name (default:localhost)--port: port (default:27017)--root-data-dir: root directory for input data (default:./test/data)
You can load the configuration parameters directly in python using DBGen.
The method load_cfg will return an object of class argparse.Namespace:
import dbgen
configs = dbgen.load_cfg()
The configs object will be required by other DBGen methods to update the database.
DBGen tables¶
The database schema of DBGen is composed of 5 tables:
Species: the basic unit of classification and a taxonomic rank of an organismDataset: an homogeneous collection of organisms’ samplesSample: collected individualsPhenotype: observable characteristics of a sampleResult: output result of bioinformatic tools
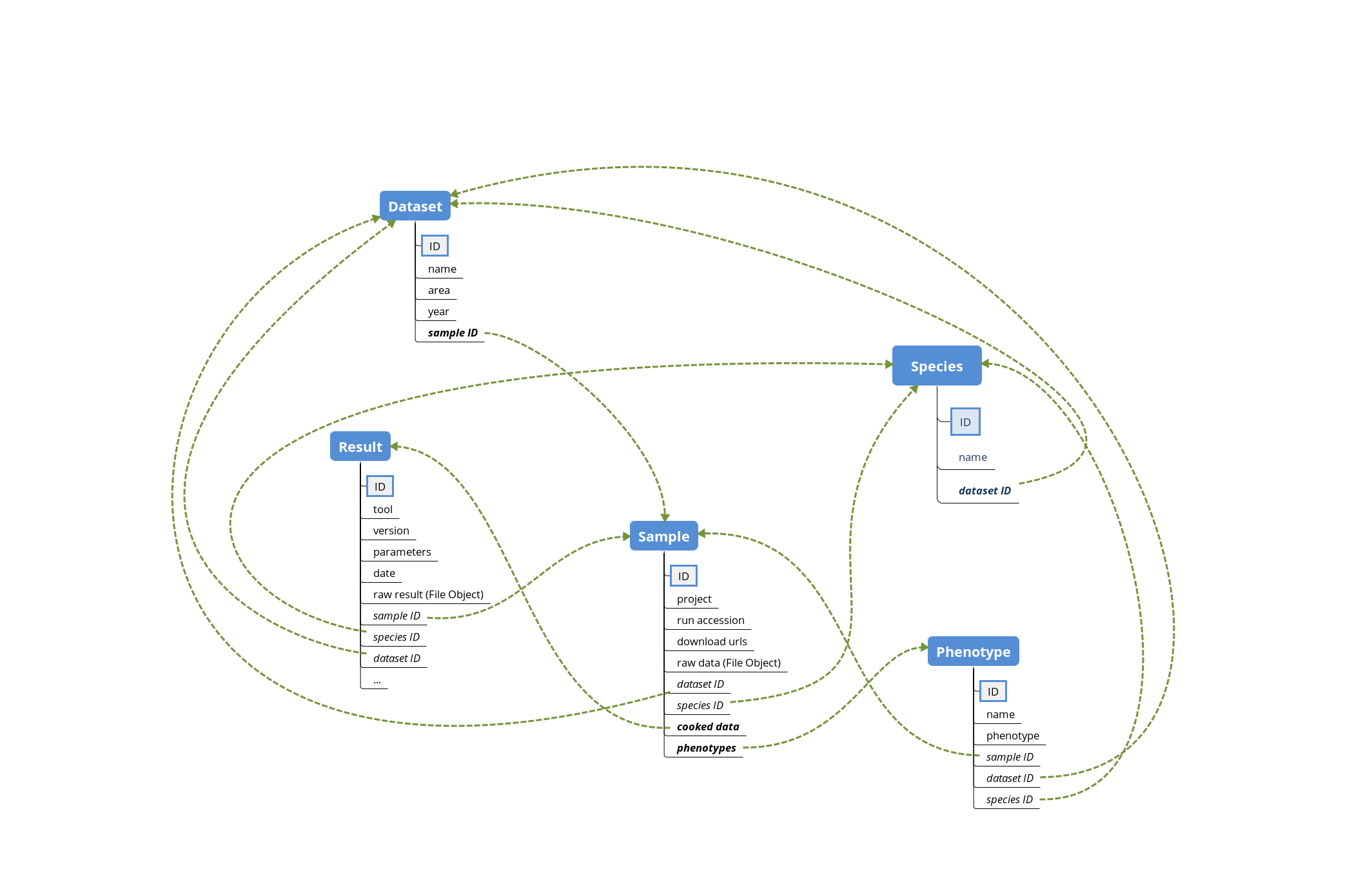
Workflow¶
The typical use of DBGen consists of 4 elements:
load sample data (with the corresponding phenotypes) from source files
download sample genotype (e.g. fastq files)
save the results (raw files) of bioinformatic analyzes (e.g. VCF files)
query the database
Load data¶
Once you have loaded the configuration parameters and started the backend database service, you are ready to upload data into DBGen.
In order to simplify the insertion of new information, DBGen is designed to scan a user-defined
directory looking for new data. The pre-defined directory is ./test/data. You can change
the default directory using the command line parameter --root-data-dir.
In order to work properly, DBGen requires a specific tree structure under the root data directory:
root-data-dir
|
+-- <species name>
| +-- <year>_<publicationName>.txt
| +-- <year>_<publicationName>.txt
| +-- ...
|
+-- <species name>
| +-- <year>_<publicationName>.txt
| +-- ...
+-- ...
Each source file must be a TSV (tab-separated values) file with the following columns:
Project name |
URLs |
Run accession |
Phenotype A |
Phenotype B |
… |
PRJNA497094 |
ftp.baz/SRR8074810_1.fastq.gz;ftp.baz/SRR8074810_2.fastq.gz |
SRR8074810 |
R |
S |
|
PRJNA497094 |
ftp.baz/SRR8074811_1.fastq.gz;ftp.baz/SRR8074811_2.fastq.gz |
SRR8074811 |
R |
S |
Download sample genotype¶
DBGen provides a simple method to download samples’ genotype. You just need to specify the name of the species and/or the name of the dataset you are interested in.
dbgen.Sample.download_raw_data(species_name="<species name>", dataset_name="<year>_<publicationName>")
Save bioinformatic results¶
Once you have obtained the results a bioinformatic pipeline, you can
easily import the results inside DBGen using the save_result method.
dbgen.Sample.save_result(sample_id=sample_primary_key,
tool_name="<tool name>",
version="<tool version>",
date="<current date>",
parameters="<tool parameters>",
raw_result_path="</path/to/result/file>")
Query the database¶
There are 6 basic methods available in DBGen to query the database:
get_phenotype_names: get the set of phenotype namesget_tool_names: get the set of bioinformatic tool namesget_download_urls: get samples’ URLs used to download their genotypeget_raw_data: get samples’ genotype (e.g. fastq files)get_phenotypes: get the observed samples’ phenotypeget_results: get the results of bioinformatic analyzes
The first two methods return a python set of strings.
The last four methods, instead, return a pandas DataFrame.
Get phenotype names¶
You can get the list of available
phenotypes using the get_phenotype_names method.
You just need to specify the name of the species and/or the name
of the dataset you are interested in.
dbgen.Phenotype.get_phenotype_names(species_name="<species name>", dataset_name="<year>_<publicationName>")
The method will return a python set of strings, i.e.:
('<phenotype name 1>', '<phenotype name 2>', '<phenotype name 3>', ...)
Get tool names¶
You can get the list of
tools used so far using the get_tool_names method.
You just need to specify the name of the species and/or the name
of the dataset you are interested in.
dbgen.Result.get_tool_names(species_name="<species name>", dataset_name="<year>_<publicationName>")
The method will return a python set of strings, i.e.:
('<tool name 1>', '<tool name 2>', '<tool name 3>', ...)
Get raw genotype¶
You can get the raw genotype
of samples using the get_raw_data method.
You just need to specify the name of the species and/or the name
of the dataset you are interested in.
dbgen.Sample.get_raw_data(species_name="<species name>", dataset_name="<year>_<publicationName>")
The method will return a pandas DataFrame, e.g.:
Sample primary key |
Project |
Run accession |
Species |
Dataset |
Genotype files |
652g6736f37719hbd |
PRJEB5225 |
ERR410034 |
Staph. Aureus |
2009_Austin |
[<RawFile>, <RawFile>] |
dug36ij3db73d8h92 |
PRJEB5225 |
ERR410035 |
Staph. Aureus |
2009_Austin |
[<RawFile>, <RawFile>] |
… |
… |
… |
… |
… |
… |
The last column will contain the raw genotype of each sample. If you want to access the file, you can use the following procedure:
results = dbgen.Sample.get_raw_data(dataset_name="2009_Austin")
# get the first genotype file of the first sample
raw_data = results.iloc[0, -1][0]
# save the file locally so that
# it can be processed by bioinformatics tools
with open(raw_data.name, "wb") as f:
raw_bytes = raw_data.file.read()
f.write(raw_bytes)
Get genotype URLs¶
You can get the list of URLs used to download the genotype
of samples using the get_download_urls method.
You just need to specify the name of the species and/or the name
of the dataset you are interested in.
dbgen.Sample.get_download_urls(species_name="<species name>", dataset_name="<year>_<publicationName>")
The method will return a pandas DataFrame, e.g.:
Sample primary key |
Project |
Run accession |
Species |
Dataset |
Genotype files |
652g6736f37719hbd |
PRJEB5225 |
ERR410034 |
Staph. Aureus |
2009_Austin |
[ftp.baz/ERR410034_1.fastq.gz, ftp.baz/ERR410034_2.fastq.gz] |
dug36ij3db73d8h92 |
PRJEB5225 |
ERR410035 |
Staph. Aureus |
2009_Austin |
[ftp.baz/ERR410035_1.fastq.gz, ftp.baz/ERR410035_2.fastq.gz] |
… |
… |
… |
… |
… |
… |
The last column will contain the list of URLs to download the genotype of each sample.
Get phenotypes¶
You can get samples’ phenotypes using the get_phenotypes method.
You just need to specify and/or:
the name of the species
the name of the dataset
plus the name of the phenotype you are interested in.
dbgen.Phenotype.get_phenotypes(species_name="<species name>",
dataset_name="<year>_<publicationName>",
phenotype_name="<name of the phenotype>")
The method will return a pandas DataFrame, e.g.:
Sample primary key |
Species |
Dataset |
Mupirocin |
652g6736f37719hbd |
Staph. Aureus |
2009_Austin |
R |
dug36ij3db73d8h92 |
Staph. Aureus |
2009_Austin |
S |
… |
… |
… |
… |
The last column will contain the observed phenotype for each sample.
Get results¶
You can get the results of bioinformatic pipelines
using the get_results method.
You just need to specify the name of the species and/or the name
of the dataset you are interested in.
dbgen.Sample.get_download_urls(species_name="<species name>",
dataset_name="<year>_<publicationName>")
The method will return a pandas DataFrame, e.g.:
Sample primary key |
Species |
Dataset |
Tool |
Version |
Parameters |
Result |
652g6736f37719hbd |
Staph. Aureus |
2009_Austin |
AMRFinder+ |
1.0.0 |
-l 20 |
<File Object> |
dug36ij3db73d8h92 |
Staph. Aureus |
2009_Austin |
BLASTn |
0.8.0 |
-p 0.2 |
<File Object> |
dug36ij3db73d8h92 |
Staph. Aureus |
2009_Austin |
AMRFinder+ |
1.1.2 |
-l 30 -k 24 |
<File Object> |
… |
… |
… |
… |
… |
… |
… |
The last column will contain a file object containing the result of each bioinformatic tool.
Examples¶
Some working examples are presented in this section. Consider the following directory structure.
./test/data
|
+-- species1
| +-- 2010_AuthorName1.txt
|
+-- species2
+-- 2009_AuthorName3.txt
+-- 2011_AuthorName3.txt
import os
import sys
import dbgen
def test_dbgen():
# load configuration and connect to the backend database
configs = dbgen.load_cfg()
dbgen.start_db(configs)
dbgen.connect_db(configs)
# load source data
dbgen.import_data(configs)
# some queries
s1 = dbgen.Sample.get_raw_data(species_name="species2", dataset_name="2009_AuthorName3")
s2 = dbgen.Sample.get_raw_data(dataset_name="2009_AuthorName3")
s3 = dbgen.Sample.get_raw_data(species_name="species2")
u1 = dbgen.Sample.get_download_urls(dataset_name="2009_AuthorName3")
u2 = dbgen.Sample.get_download_urls(species_name="species2")
u3 = dbgen.Sample.get_download_urls(species_name="species2", dataset_name="2009_AuthorName3")
p0 = dbgen.Phenotype.get_phenotype_names(species_name="species2")
p1 = dbgen.Phenotype.get_phenotypes(species_name="species2", phenotype_name="Mupirocin")
p2 = dbgen.Phenotype.get_phenotypes(dataset_name="2009_AuthorName3", phenotype_name="Mupirocin")
p3 = dbgen.Phenotype.get_phenotypes(species_name="species2",
dataset_name="2009_AuthorName3",
phenotype_name="Mupirocin")
# download samples' genotype
dbgen.Sample.download_raw_data(species_name="species2", dataset_name="2009_AuthorName3")
# get raw genotype
s1 = dbgen.Sample.get_raw_data(species_name="species2", dataset_name="2009_AuthorName3")
s2 = dbgen.Sample.get_raw_data(dataset_name="2009_AuthorName3")
s3 = dbgen.Sample.get_raw_data(species_name="species2")
# save a local genotype file
raw_data = s1.iloc[0, -1][1]
with open(raw_data.name, "wb") as f:
raw_bytes = raw_data.file.read()
f.write(raw_bytes)
# pretend to run a bioinformatic pipeline
# on some samples and save the results
# into DBGen
root_path = "./test/db/cooked/"
if not os.path.exists(root_path):
os.makedirs(root_path)
for k, v in s1.iterrows():
tool_name = "AMRFinder+"
version = "0.0.1"
date = "2019-09-02"
parameters = "-c 20 -v 39"
file_path = os.path.join(root_path, "testfile_%s.txt" % v["run accession"])
raw_result = open(file_path, "w")
raw_result.write("Hello %s" % v["run accession"])
raw_result.write("This is our new text file")
raw_result.write("and this is another line.")
raw_result.write("Why? Because we can.")
raw_result.close()
raw_result_path = os.path.abspath(file_path)
dbgen.Sample.save_result(k, tool_name, version,
date, parameters, raw_result_path)
# query the result table
res0 = dbgen.Result.get_results(species_name="species2", dataset_name="2009_AuthorName3")
res1 = dbgen.Result.get_results(species_name="species2")
res2 = dbgen.Result.get_results(dataset_name="2009_AuthorName3")
# shutdown the database backend
dbgen.shutdown_db(configs)
return